前言
在数据管理部分,我们应用summarizeOverlaps 来管理RNAseqData.HNRNPC.bam.chr14中的BAM文件。现在我们再次来操作一下。
使用SummarizedExperiment来管理BAM文件
|
|
hnse 是RangedSummarizedExperiment 类的一实例。这个类就类似于 ExpressionSet ,不过有着更多的内容来管理元数据,它的流程如下所示:
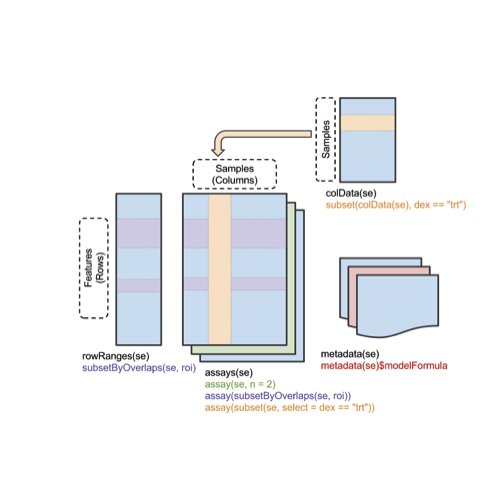
有效地使用SummarizedExperiment实例涉及学习它的一些方法。为了获取HNRNPC基因的读长/区域(read/region),我们可以使用assay方法,如下所示:
|
|
以上就是最基本的结果表示方法。列名则是样本标识符,但是有关区域检查的一些信息则已经丢失。
SummarizedExperiment中的元数据
hnse 对象还有一些其它的信息,如下所示:
|
|
我们还可以进一步分析差异信息,从而输出更多,更广泛的信息。
通过添加输入信息来更有效地生成SummarizedExperiment
使用元数据定义感兴趣的区域
We have seen that it is sufficient to define a single GRanges to drive summarizeOverlaps over a set of BAM files. We’d like to preserve more metadata about the regions examined. We’ll use the TxDb infrastructure, to be described in more detail later, to get a structure defining gene regions on chr14. We’ll also use the Homo.sapiens annotation package to add gene symbols.
|
|
定义BAM文件的样本信息
现在我们有三个样本,一个用于控制,两个用于敲低。我们使用GenomicFiles来绑定样本信息的元数据,如下所示:
|
|
比较读长覆盖区,保留元数据
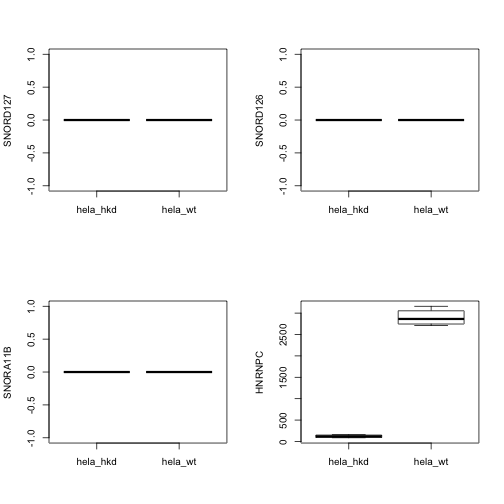
我们来查看5个基因,其中就包括HNRNPC。当计算后,我们会将样本信息再绑定回结果,如下所示:
|
|
从上面我们可以看出,行标识符是随计数矩阵一起出现的,现在我们绘制出这几个基因的箱线图,如下所示:
|
|
从ExpressionSet提取数据
从ExpressionSet中提取数据很容易,但是还需要基于基因组范围查询的阵列探针的子集,如下所示:
|
|
总结
RangedSummarizedExperiment类实例化了一些Bioconductor数据结构设计的一些关键原则:
- 基于样本特征的数据分析和元数据分析,并将它们以某种方式绑定在一起。
- 类似于矩阵的子集直接用于分析和样本数据分析。
- 基于范围的子集设置适用于可通过基因组坐标寻址的分析。
- 可以在
mcol中提供关键分析特征的任意元数据(rowRanges(se))。 - 可以通过
metadata(se)<-来添加任何元数据。
在后面的内容里我们还将会了解更多的关于SummarizedExperiment改造的一些数据结构,从而用于专门用于RNA-seq的多个阶段处理和数据分析。